Rick Young is a professor of biology at MIT who studies RNA that is transcribed from the part of the genome that does not code for proteins, known as non-coding DNA. This part of the genome was once referred to as ‘junk DNA,’ which gives you a sense of what many thought of its value. Scientists were startled to discover that it makes up 98% of the human genome, which triggered a quest to find its functions.
In this conversation, Rick Young chats with Hanne Winarsky from Bio Eats World and a16z general partner Jorge Conde, who leads investments at the intersection of biology, computer science, and engineering. Before joining a16z, Conde was Chief Strategy Officer at Syros Pharmaceuticals and co-founded the genomics interpretation company Knome.
The conversation covers what we’ve learned about that 98% of the genome we thought was junk. Turns out, it has diverse jobs ranging from hiding away the evidence of ancient viral infections to making every face look unique. They also discuss its massive but still poorly understood role in disease, and how studying junk DNA led to the discovery of a gene on/off switch that no one expected.
Note: this conversation originally was originally published as an episode of Bio Eats World. You can listen to that episode here.
HANNE WINARSKY: We’re here to talk today about what is called junk DNA. Can we start with just a simple definition?
RICK YOUNG: That’s about a half-century old term. Scientists knew about portions of the genome that don’t encode proteins, and they theorized that this was junk. We knew some of it was just the remnants of ancient viral invasions of the genome. But that phrase, junk DNA, has haunted us.
HANNE: So what is the term that you’re trying to use instead? The dark matter of DNA that we’re understanding more about every day?
RICK: Non-coding DNA.
HANNE: Why did they think of it as detritus? You’ve mentioned some of it was leftover old virus bits. But why wasn’t it just a mystery from the beginning?
RICK: Because throughout biological history, there was this debate over what was the genetic material, and initially, it was thought to be protein. But once it became clear that protein was the machinery and DNA was the blueprint for the machinery, people got busy on the machinery because defects in the machinery cause disease. But then it turned out that only 2% of the genome is encoding the amino acids for proteins. The vast majority, 98%, does not. And in 2000, when scientists of the Human Genome Project presented the human genome sequence, that data confirmed that 98% of our 3.2 billion bases don’t encode proteins.
Each gene has that remarkable capability of taking bits and pieces of segments of the protein that it will encode and arranging it so that the product that you get in one cell might be a little faster working, or in another cell may actually go into a different compartment to do a different job.
JORGE CONDE: What were the initial estimates to how many genes would be encoded in those 3.2 billion base pairs?
RICK: We settled on about 100,000. We just assumed that the more complex we are, the bigger the genome, and the larger the number of genes. There was a bit of a shock when we realized that we and insects have about the same number of genes.
JORGE: Fewer genes than we anticipated encoding for what we consider to be an incredibly complex organism, right?
HANNE: That is a bit of a shock.
Same source code, different programs
JORGE: A thing we all learned in high school is that DNA codes for RNA, RNA codes for amino acids, and amino acids give us proteins, right? That’s the central dogma of modern biology.
RICK: Yep. One of the big reasons why people were quick to ascribe the title ‘junk DNA’ to that 98% of the genome that does not code for proteins is because it was believed, in large part, that the business end of the genome was to make proteins.
JORGE: So when did geneticists start to get an inkling that junk DNA may be more than junk?
RICK: [It started with] the realization that you could account for the additional complexity in human beings versus insects by a tremendous amount of alternative splicing. That’s where you have, for a single gene, a large RNA that’s made, but it gets spliced differently in one cell versus another cell. In other words, different portions of the gene end up in the RNA molecule that’s going to specify the protein. So the protein is a little different.
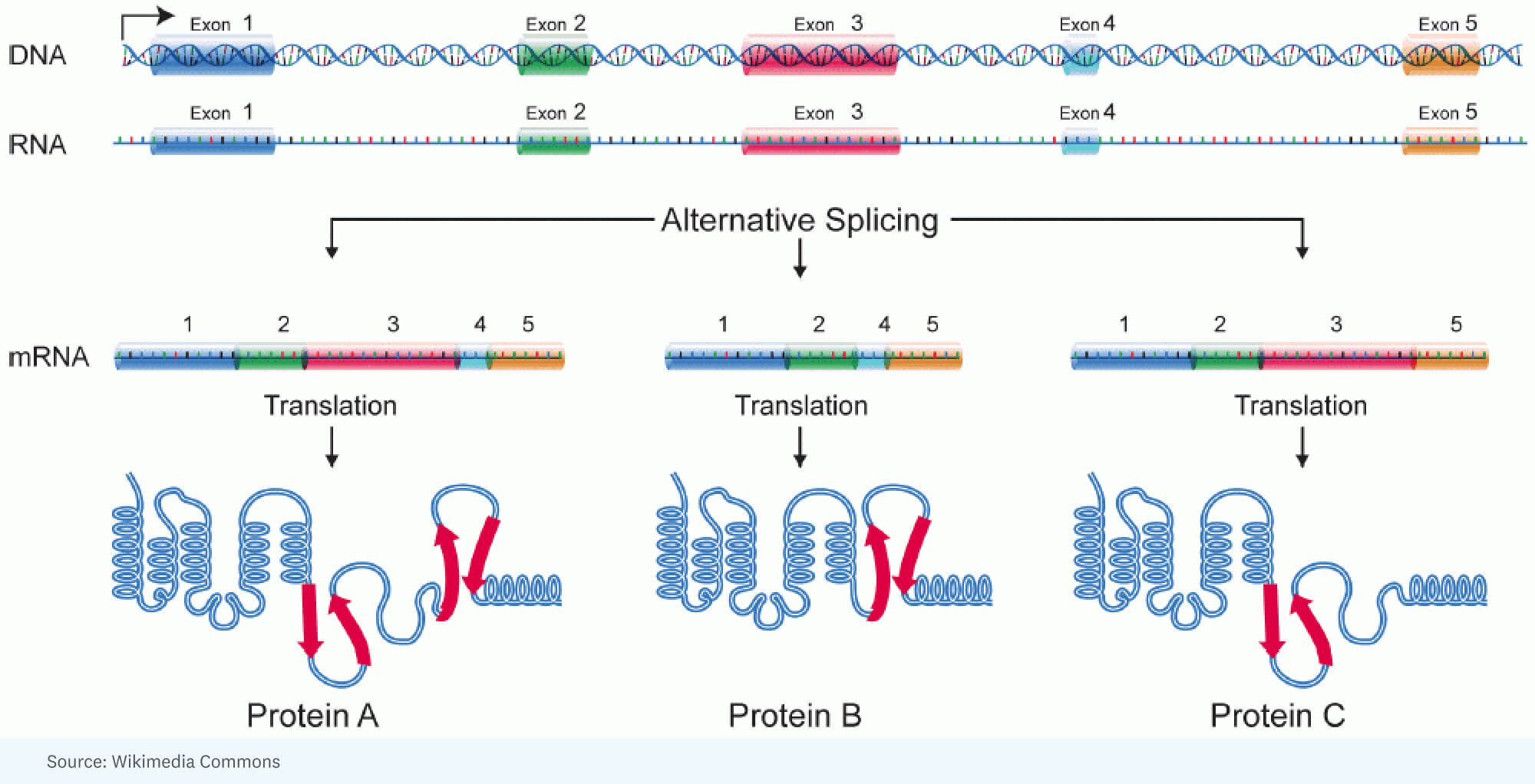
National Human Genome Research Institute, Public domain, via Wikimedia Commons
HANNE: That sounds like a kaleidoscope a little bit with light hitting it differently, you get different colors, different angles.
RICK: Well, and that’s an interesting analogy. I think a better analogy is when you have these Legos, and you can make a machine, but you can make it in so many different ways, so many different structures, colors. Each gene has that remarkable capability of taking bits and pieces of segments of the protein that it will encode and arranging it so that the product that you get in one cell might be a little faster working, or in another cell may actually go into a different compartment to do a different job.
JORGE: Every single cell in a given human has approximately the same genome. Yet that same genome gives rise to an incredibly diverse array of different cell types. And so to the extent that we are going to make an analogy, each cell type is running a different program off of the same source code.
RICK: That’s right.
The functions of the 98%
JORGE: You don’t need to be an expert to look at different cell types and see how varied they can be, right? A neuron looks very, very, very different and functions very, very differently than, say, a muscle cell. What determines the program, the genetic program that a cell chooses to run? What makes a muscle cell a muscle cell, and what makes a neuron a neuron?
RICK: So we started out with DNA makes RNA and [RNA] makes protein. That’s the central dogma. But about half a century ago, scientists began making the argument that in fact RNA began to create various kinds of functions all on its own. And it turns out that RNA actually has some of the activity at the earliest stages of development.
When the sperm meets the egg, it’s the mother’s RNA that she puts into that egg. There are RNA molecules that are doing this. It turns out antibiotics that we use routinely bind to the RNA. So the RNA has some pretty important roles there. That changed the way people think. Then, as we started to think about junk DNA, that’s the part of DNA that’s not encoding protein. Well, what if the world is based on RNA and not protein, at least at the beginning? And so now we understand that a huge fraction of what we call junk DNA, or what we used to call junk DNA, is not junk. It’s highly functional. And most of it makes RNA.
So your goal in programming any one cell is to use just that specific set of sequences that will tune each of that common set of genes to the level you want. . . .Our problem is we don’t actually know the program.
HANNE: Wow. Can you do a bit of a lay of the land of where we are in understanding the noncoding part of the DNA? You know, what is our current understanding of all the different possibilities there?
RICK: Only 2% of our genome is encoding these amino acid sequences that go into proteins. So what’s on our accountant’s ledger for what the rest does?
About half of our genome is what we call heterochromatin. That’s where you get the products of ancient viral invasions. Ancient retroviruses invaded, and then were turned into DNA, and they were inserted into the genome. So that actually is a means that we’ve had throughout our evolutionary history to hide away sequences that we don’t want to deal with. And it remains silent in our genome with an important exception.
The other half is where all the active protein coding genes are, and where all the active noncoding genes are. So, what does it do? It has a long list of regulatory functions, but I’ll simplify it into three.
One of its functions is chromosome maintenance. So, those are the places where DNA replication occurs. They’re the sites in our genome that are responsible for folding it up because it’s a 2-meter long polymer. It’s got to get folded up into a couple micron diameter nucleus.
The second regulatory region is all these things that are responsible for gene regulation. Probably much more of the genome specifies regulatory features for gene expression than specifies genes themselves. And that is because each cell uses a different regulatory region for each gene.
HANNE: It’s so interesting, it sounds to me a little bit almost like there’s the closet with the shelves on it of things we need to put in the closet for a little while, and then there’s the infrastructure closet.
Why is it important to focus so much on this? Because that’s where over 75% of all disease-associated genetic variation occurs.
RICK: Yes. Basically, what you have is a common set of genes in every cell, both coding and noncoding. And you have elements, you have actual sequences that are operating only in specific cell types. So your goal in programming any one cell is to use just that specific set of sequences that will tune each of that common set of genes to the level you want. So you’re playing an amazing musical instrument of 20,000 protein coding genes, and about the same number of noncoding genes. You’re doing that through specific sequences. Our problem is we don’t actually know the program.
Teasing out the regulatory program
HANNE: So how do you begin to suss it out? What are the hints that you’re following when you’re starting to try to understand this program?
RICK: The hints are that the regulatory regions for each gene in a cell display themselves. They tell you. And you can use various technologies that very quickly tell you across the entire genome, in a particular cell type, let’s say in a motor neuron, what are all the regulatory regions that are on in that cell. You can even see where the rheostat is set for each of those genes. That’s where rapid sequencing has given us these capabilities to simultaneously deduce all of the active elements for genes, both coding and noncoding in the genome of a particular cell type.
Our problem at the moment is you have to do this pretty much one cell type at a time, and we have many, many hundreds of cell types. Sometimes it’s hard to actually see a particular cell without contaminating with other cells, because all our tissues really are combinations of multiple cell types.
JORGE: Is it worth arguing by analogy if we said that given that every cell has the entire genome, every cell has the entire songbook, specific cell types choose to play specific symphonies, and the machinery that helps regulate the genome is essentially the conductor of the orchestra? That machinery is the conductor that determines what songs to play, what notes to hit, at what volume to hit them, at what tempo, etc. Is that a reasonable analogy to understanding the regulatory function of the genome?
RICK: It is in the sense that it’s easy to see then what the output would be. But what’s more challenging is, who writes all the notes? Who’s the composer that put all those notes in there, and got it all right? The composer turns out to be, for most of our cells and most of our genes, these protein molecules called transcription factors, whose job it is to bind to the regulatory elements of genes, and give them a rheostat setting.
Now, there’s an interesting wrinkle in this because at those sites where those transcription factors bind, we call them an enhancer. At those enhancer sites, there’s also always an RNA being made from that site where they’re bound. We have only recently come to understand that that RNA plays important roles in regulation. Just to amplify that: the way your iPhone recognizes your face is because the enhancers that control cranial facial structure genes vary in each human being.
What you have now here is this triumvirate. You have the DNA sequence. It’s recognized specifically by the composing molecule, the transcription factor, but it needs this third piece, this RNA molecule. So the DNA, RNA, and protein actually work together at those regulatory regions. And why is it important to focus so much on this? Because that’s where over 75% of all disease-associated genetic variation occurs.
HANNE: Not to get too musically nerdy, but it almost sounds like a chord, right? The three-note structure all playing together to create something larger.
RICK: That’s right.
The programmers
JORGE: One of the most cutting edge areas of biology is our increasing ability to try to understand some of the governing laws of how cell programs are determined, how cell fate is determined. For me one of the fascinating leaps forward in our understanding, came from the work that Yamanaka did, for which he was awarded the Nobel Prize, demonstrating that you could reprogram cell types by just exposing cells to a very small handful of specific transcription factors.
HANNE: Can you describe why it was exactly that it was such a breakthrough for the field?
RICK: I had a tiny bit role in that movie. It turns out that although that’s a very large number, a small number of transcription factors can identify all the regulatory elements that are essential for that cell’s identity. And Yamanaka proved this to us by showing that only four of these factors could be used to program any human cell, or any male cell into the equivalent of an embryonic stem cell.
One way to think about this is, if the song is too bad, the organism doesn’t live. But if it’s just a bit off, you grow up, you become an adult, and then you acquire all these various diseases as we get older.
JORGE: And that’s amazing, right? Because that would suggest that the system is somehow designed where incredible complexity is drawn from what sounds like simplicity. Four transcription factors determining all the complex cascade of events that govern different cell types.
Some of the work you have done has demonstrated that these master transcription factors essentially set up the equivalent of circuits that control the genes that are necessary for a cell to establish and maintain its state. Can you describe what you mean by gene control circuits?
RICK: There are two cool elements to the gene control circuits. One is, when a master regulator finds these enhancers and causes the expression of its target genes, that’s a part of the circuitry, that’s the output. The other element that is so cool is that the master transcription factors also regulate their own expression. So there’s a feedback loop. Like, you would have an electrical diagram where you have the masters controlling their own expression from their own genes, and then binding to and controlling expression of a target set of genes.
JORGE: That’s pretty wild. It’s almost like a circular reference, where transcription factors are protein, that protein is made from DNA, encoded in a gene. Transcription factors are part of the machinery that helps the expression in transcription of genes. And so therefore, you’re saying transcription factors–the protein–help regulate the expression of the genes that make the transcription factors.
HANNE: Yeah. There’s a mental image of this entire symphony of all these little cells, you know, singing out all these different textures.
The regulatory genome and disease
HANNE: What does it change when we begin to understand how this all functions? What can we do with this knowledge?
RICK: These sites where these master transcription factors are driving each cell’s identity is where most of human variation is that causes disease. Over 75% of disease-associated variation occurs in these enhancer elements that are driving the key genes.
JORGE: Okay. So that’s wild, right? When we think about mutations causing or contributing to disease, we normally think about a mutation that occurs within a gene that affects the protein, somehow breaks the protein, and that gives rise to disease.
HANNE: Right.
JORGE: But you’re saying is that in 75% of the cases, that mutation is actually happening outside of the genes, it’s happening in this noncoding region of the genome. If the gene is the song, it’s not that the song is being misplayed, it’s that it might be played too loud, or too soft, or too slowly, or too quickly, but that’s what drives a lot of disease.
RICK: In fact, one way to think about this is, if the song is too bad, the organism doesn’t live. But if it’s just a bit off, you grow up, you become an adult, and then you acquire all these various diseases as we get older.
For the first time, we have all these models for how you set up the apparatus and make it work.
JORGE: Not making the wrong version of the gene, but getting the wrong dosage of the gene. Too much or too little.
RICK: That’s correct. How do you find therapies that deal with this? How do you selectively tune up or tune down the gene? In principle, we can do that in a lot of ways, and we can do that with gene therapy. We can do that with CRISPR gene editing. But the most important thing I think we’ve discovered in the last few years is that each of these gene regulatory elements has an RNA. The RNA is functional. It’s a rheostat that helps tune the output of that gene. There are now many ways that you can drug RNAs. We’ve got ASOs (antisense oligonucleotides), such as Spinraza for spinal muscular atrophy. We’ve got RNA interference. We’ve got some new small molecule drugs on the horizon. If you could think about ways of now programming a drug, a synthetic RNA, to regulate the regulator RNA, the regulatory RNA, you have the principal way of tuning any one gene in any cell where that cell can gain access to that drug.
HANNE: So it’s not just a whole different understanding of how disease emerges. But it’s a whole different understanding of how we could potentially treat disease.
RICK: Exactly. In principle, we now have a programmable way of developing a drug that tunes any one gene of interest. At this moment in time, people are simply programming synthetic RNA molecules to produce a vaccine for this pandemic. One that is as good a result as you could ever expect for a vaccine.
JORGE: When we think about the applications of technology in biology, we’re usually trying to do one of two things. We’re either trying to interrogate biology very deeply, and understand it, increasing levels of its complexity, or we’re trying to intervene. We increasingly are able to interrogate biology at a very, very deep level so we understand the governing laws or the rules by how cells are regulated. And we have that, we have increasingly sophisticated tools, like these programmable modalities of medicine, where we can target RNA, very, very specifically. This will sort of be this virtuous cycle between our ability to interrogate biology and then intervene in increasingly sophisticated ways. And I think that’s one of the most exciting aspects of where we find ourselves today in this field.
RICK: I agree with you. We now are developing such a deep understanding of the multiple layers of complexity, that we can come up with therapeutic hypotheses that we’ve not seen before. We can do them with a speed that we never conceived of only a few years ago. That temporal distance between a basic discovery and the therapy that went into people 10 years ago was 14 years on average. Now, it’s conceivable to think of developing a therapeutic hypothesis based on basic science, and a therapy that reaches a patient in nine months. We’re seeing that with this new vaccine.
HANNE: So, changing not just how we understand disease emerging, how we treat it, but also how we do the science itself, and then how fast the science can happen and turn into clinical reality for patients.
RNA as compartmentalizer
RICK: Exactly. But now there’s icing on the cake because, classically, we’ve thought about pharmacology in two ways. One was the effect of the drug on the individual. The other was the effect of the individual on the drug. And in this latter segment, you’re worried about distribution of the drug, what tissues it goes to, what tissues it’s not available to. Because we just assume once a drug gets into a cell, it diffuses through the cell and finds its target. We have membrane-bound compartments, which we’ve known about for a century.
JORGE: Which was always the question of the cell permeability, right? Can it cross the membrane?
RICK: Yes. Can it cross a membrane, and does it get into the nucleus or not? But we’ve only come to understand in the last decade that there are also many non-membrane bodies in cells called biomolecular condensates because it’s thought that one reason that these bodies form is they condense much like water condenses into a dewdrop. But what has been so profound about this understanding is that these condensates compartmentalize proteins, DNA, RNA for specific functions. And so now we’ve come to understand that you can segregate the 5 to 10 billion protein and RNA molecules in a cell into various compartments where they function with their buddies.
HANNE: Huh.
JORGE: Are we leaving the realm of biology and entering the realm of physics?
RICK: We have done exactly that because phase separation is thought to be the driving force. That is a physical phenomenon described by math.
HANNE: Wow.
RICK: Now, we’ve learned the most effective chemotherapeutic drugs are concentrating inside the compartments where their targets live. They’re concentrating 600-fold over the rest of the cell, so they have on-target activity on oncogenes that is 600 times what we expected. This not only tells us that there are brand new insights that are important in drug discovery and development for the future, but it makes us want to better understand what these condensates do.
Here is what I mean by the icing on the cake. What we’ve come to realize is that these condensate compartments that are functionalizing the cell in such important ways are regulated by RNA. Their formation can be stimulated by RNA. If you produce too much RNA, you bring the rheostat up to 11, it will dissolve a condensate. So, suddenly, we realize that the RNA output at any site inside a cell can tune the function of anything by enhancing or dissolving those condensates where that function is occurring. And that is, I think, profound because it is another way that a programmable RNA, a synthetic RNA molecule, might be employed to tune the function of a cell that’s become dysfunctional. For the first time, we have all these models for how you set up the apparatus and make it work.
HANNE: Another knob to dial.
RICK: But then how do you turn it off? It turns out that when you make that long RNA, that’s just a big string of negative charges, and it dissolves the condensate and shuts the gene down. That is how genes get regulated. You tune up the condensate with an RNA, then you shut it down with the RNA product that’s made when the gene gets fully transcribed.
HANNE: Super cool. So an off and on switch, really.
RICK: It’s an off/on switch no one anticipated. And it means, once again, if you have a programmable drug, you have a new way of targeting cellular functions that are dysfunctional, a new solution for a therapeutic problem.
JORGE: One man’s junk DNA is another man’s sophisticated genome regulatory machinery.
HANNE: Or every man’s.
